- Homme de 42 ans, porteur d’une maladie d’Addison
- Troubles de la marche depuis 1 an
- ATCD familiaux : 2 demi-frères avec troubles de la marche (1 avec épilepsie)
- Examen clinique :
- Paraparésie spastique
- Hypoesthésie distale symétrique MI
- Syndrome cérébelleux
- Poursuite saccadique
- IRM cérébrale (FLAIR, et T1 Gd)
Quel est votre diagnostic ?
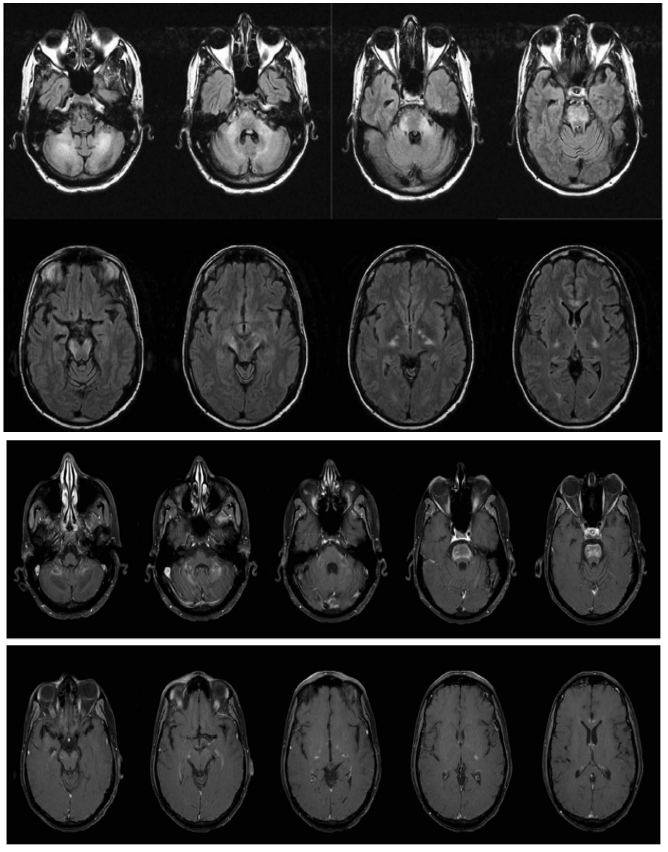
L’adrénoleucodystrophie (ALD)
C’est une maladie génétique récessive liée à l’X qui atteint principalement la substance blanche, les corticosurrénales et les testicules.
Elle est responsable d’un défaut du catabolisme dans le peroxysome des acides gras à très longue chaîne. La maladie comporte une grande variabilité phénotypique.
La forme cérébrale de l’enfant est la plus fréquente et représente environ la moitié des cas ; elle survient après un développement psychomoteur normal mais entraîne une détérioration cérébrale rapidement progressive conduisant à un état végétatif en deux ans.
La seconde forme la plus fréquente est l’adrénomyéloneuropthie de l’adulte (25 %), secondaire à la démyélinisation des cordons postérieurs et antérieurs de la moelle tandis que la substance blanche cérébrale est respectée. Les symptômes neurologiques apparaissent en moyenne à l’âge de 30 ans sous la forme d’une paraparésie spastique et d’une neuropathie périphérique modérée.
Les autres formes, plus rares, sont l’atteinte cérébrale de l’adolescent (5 %), de l’adulte (3 %)
et une insuffisance surrénalienne isolée (10 %).
De plus, certains sujets sont porteurs de l’anomalie génétique mais restent asymptomatiques (8 %).
On classera à part les sujets hétérozygotes symptomatiques : quinze à vingt pour cent des femmes hétérozygotes pour l’ALD peuvent développer une paraparésie spastique progressive, une neuropathie périphérique, des troubles sphinctériens se manifestant en moyenne à 43 ans (+/– 11 ans).
Référence : Desloques et al. 2006